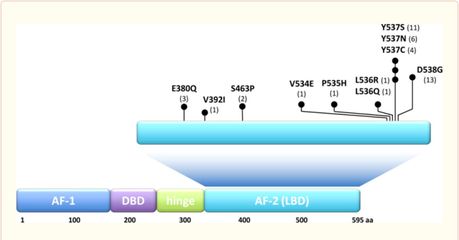
Our group has identified the ESR1 ligand binding activating mutations as the most common genomic mechanism of acquired endocrine resistance in metastatic hormone receptor positive breast cancer. These mutations lead to constitutive activity resulting in resistance to aromatase inhibitors. The ESR1 mutations also lead to decreased affinity to tamoxifen and fulvestrant and relative resistance to these endocrine treatments.
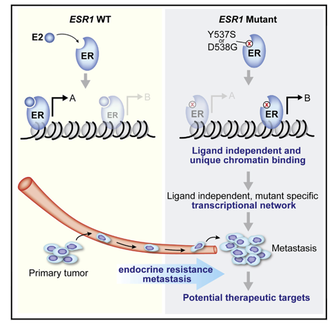
We have generated cell lines and patient derived xenograft models harboring the ESR1 ligand binding mutations. Through epigenetic analysis and functional in vivo validation studies we showed that the ESR1 mutations have neomorphic properties that promote drug resistance and metastases. We are currently investigating approaches to leverage the ESR1 mutations for therapeutic purposes.
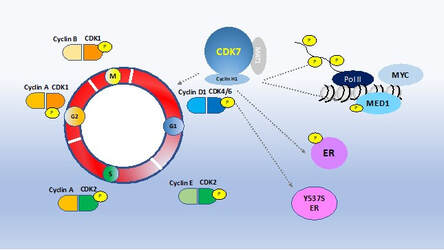
Our genome-wide CRISPR knock-out screen has identified CDK7 as an essential gene in hormone receptor positive breast cancer models including models that harbor the ESR1 mutation and models of resistance to CDK4/6 inhibitors. Currently there are a number of CDK7 inhibitors in clinical development. CDK7 is a master regulator of transcription and of the cell cycle. CDK7 also phosphorylates ER. Since CDK7 has multiple key roles in cancer, we are investigating the mechanism of action of the CDK7 inhibitors in hormone receptor positive breast cancer. We are also investigating mechanisms of resistance to CDK7 inhibitors and potential drug combinations to enhance the activity of CDK7 inhibitors.
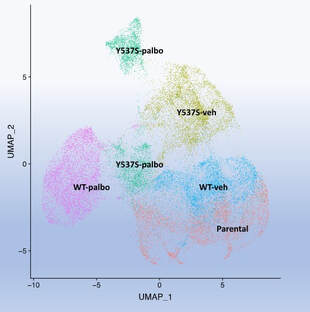
CDK4/6 inhibitors have changed the treatment paradigm in metastatic hormone receptor positive breast cancer. This class of drugs has significantly increased progression free and overall survival in metastatic hormone receptor positive breast cancer. However, about ten percent of patients have de novo resistance and ultimately all patients will develop resistance. Pre-clinical and clinical studies have identified multiple mechanisms of resistance. The Y537S ER mutation is enriched during the acquisition of resistance to CDK4/6 inhibitor treatment in combination with endocrine treatment. Given these observations we have been studying the clonal dynamics during the acquisition of resistance to CDK4/6 inhibitor and the clonal architecture of the resistant cells in the presence of WT and mutant ER.
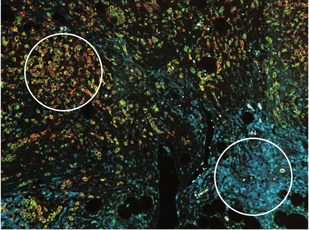
The tumor immune microenvironment plays a key role in cancer and is now being exploited for cancer treatments. Hormone receptor positive breast cancers are considered ‘cold’ cancers with low immune cell infiltrates. We are studying the effects of estrogen signaling on the tumor immune micro-environment and investigating mechanisms to enhance the immune anti-tumor activity in breast cancer. For these studies we are employing clinical tissue samples and new co-culturing systems.
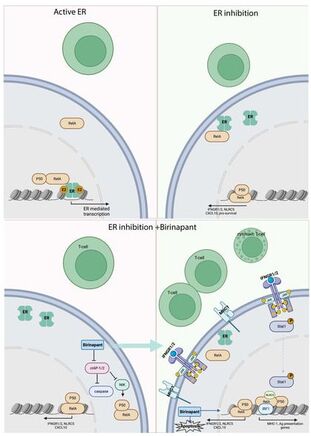
In the primary breast cancers from the neoadjuvant clinical trial we found that endocrine treatment increases the expression of B2-microglobulin, the non-polymorphic subunit of the MHC-I complex. We followed up on this clinical finding in the lab and found that ER blockade is required foe RelA genome binding, which in turn increases immune related signaling and results in enhanced response to IFN-g. In addition, we found that SMAC mimetic increase NFKB signaling in ER+ breast cancer and enhance the immunogenicity while also leading to increased cell death. These results indicate that ER blockade is important to be added to immune therapies in ER+ breast ca and that the combination of SMAC mimetics to ET can increase cell death and tumor response and can potentiate the response to immune therapies.